In fact, copykat is only unsatisfactory when judging by the algorithm, but the visualization is still that the naked eye can clearly distinguish diploid normal cells and aneuploid cancer cells, so we want to see what specific improvements can be made to bypass this bug. The first choice is that we split all epithelial cells according to patients, Get the separate folder for each patient as shown below and the expFile.txt under each folder!
81M 12 2 10:09 S11/expFile.txt 123M 12 2 10:22 S21/expFile.txt 84M 12 2 10:41 S47/expFile.txt 75M 12 2 10:54 S50/expFile.txt 78M 12 2 11:05 S53/expFile.txt 95M 12 2 11:20 S56/expFile.txt 95M 12 2 11:34 S57/expFile.txt 78M 12 2 11:49 S58/expFile.txt 107M 12 2 12:01 S63/expFile.txt 77M 12 2 12:17 S65/expFile.txt 87M 12 2 12:29 S66/expFile.txt 87M 12 2 12:42 S69/expFile.txt 79M 12 2 12:56 S71/expFile.txt 75M 12 2 13:08 S72/expFile.txt 76M 12 2 13:19 S74/expFile.txt 78M 12 2 13:34 S78/expFile.txt 77M 12 2 13:45 S79/expFile.txt 77M 12 2 13:57 S81/expFile.txt 84M 12 2 14:09 S82/expFile.txt
In this way, I only need to write a simple script Step4 run copykat. R, which can save the country by parallel curves in the way of all samples, which can also be regarded as parallel computing.
This simple script Step4 run copykat. R is the following shell script:
ls -d S*|while read id;do echo $id; cd $id; nohup Rscript.exe ../scripts/step4-run-copykat.R & cd ../ done
It is easy to see the single cell level cell expression matrix of each patient. Running this copykat process takes time:
S11/nohup.out:Time difference of 54.33337 mins S21/nohup.out:Time difference of 1.20906 hours S47/nohup.out:Time difference of 54.88378 mins S50/nohup.out:Time difference of 43.44914 mins S53/nohup.out:Time difference of 51.72247 mins S56/nohup.out:Time difference of 1.020941 hours S57/nohup.out:Time difference of 1.001784 hours S58/nohup.out:Time difference of 52.24233 mins S63/nohup.out:Time difference of 1.096883 hours S65/nohup.out:Time difference of 45.49075 mins S66/nohup.out:Time difference of 56.40917 mins S69/nohup.out:Time difference of 56.40654 mins S71/nohup.out:Time difference of 52.58897 mins S72/nohup.out:Time difference of 42.06179 mins S74/nohup.out:Time difference of 47.09917 mins S78/nohup.out:Time difference of 49.3231 mins S79/nohup.out:Time difference of 50.86545 mins S81/nohup.out:Time difference of 44.29561 mins S82/nohup.out:Time difference of 45.59241 mins
Moreover, I briefly looked at the discrimination difference between diploid normal cells and aneuploid cells in these patients:
tail -n 6 S*/nohup.out
==> S11/nohup.out <==
aneuploid diploid
epi 177 0
ref-Bcells 7 389
ref-Tcells 0 478
spike-Bcells 6 235
spike-Tcells 0 289
==> S21/nohup.out <==
aneuploid diploid
epi 935 23
ref-Bcells 9 393
ref-Tcells 0 480
spike-Bcells 4 241
spike-Tcells 0 289
==> S47/nohup.out <==
aneuploid diploid
epi 195 13
ref-Bcells 9 385
ref-Tcells 0 477
spike-Bcells 6 232
spike-Tcells 0 289
==> S50/nohup.out <==
aneuploid diploid
epi 27 8
ref-Bcells 316 77
ref-Tcells 20 457
spike-Bcells 184 55
spike-Tcells 24 265
==> S53/nohup.out <==
aneuploid diploid
epi 74 27
ref-Bcells 0 393
ref-Tcells 0 477
spike-Bcells 1 238
spike-Tcells 0 289
==> S56/nohup.out <==
aneuploid diploid
epi 348 114
ref-Bcells 0 399
ref-Tcells 0 477
spike-Bcells 0 241
spike-Tcells 0 289
==> S57/nohup.out <==
aneuploid diploid
epi 104 356
ref-Bcells 0 395
ref-Tcells 0 477
spike-Bcells 0 239
spike-Tcells 0 289
==> S58/nohup.out <==
aneuploid diploid
epi 96 2
ref-Bcells 311 82
ref-Tcells 16 461
spike-Bcells 180 60
spike-Tcells 18 271
==> S63/nohup.out <==
aneuploid diploid
epi 605 80
ref-Bcells 0 400
ref-Tcells 1 477
spike-Bcells 0 243
spike-Tcells 0 289
==> S65/nohup.out <==
aneuploid diploid
epi 56 28
ref-Bcells 307 84
ref-Tcells 32 445
spike-Bcells 190 45
spike-Tcells 29 257
==> S66/nohup.out <==
aneuploid diploid
epi 266 19
ref-Bcells 16 379
ref-Tcells 0 478
spike-Bcells 9 234
spike-Tcells 2 287
==> S69/nohup.out <==
aneuploid diploid
epi 290 3
ref-Bcells 7 391
ref-Tcells 0 479
spike-Bcells 5 238
spike-Tcells 0 289
==> S71/nohup.out <==
aneuploid diploid
epi 71 52
ref-Bcells 0 393
ref-Tcells 0 477
spike-Bcells 0 240
spike-Tcells 0 289
==> S72/nohup.out <==
aneuploid diploid
epi 28 2
ref-Bcells 317 75
ref-Tcells 29 448
spike-Bcells 188 47
spike-Tcells 32 254
==> S74/nohup.out <==
aneuploid diploid
epi 48 25
ref-Bcells 0 393
ref-Tcells 0 477
spike-Bcells 1 238
spike-Tcells 0 289
==> S78/nohup.out <==
aneuploid diploid
epi 94 8
ref-Bcells 317 75
ref-Tcells 35 442
spike-Bcells 192 44
spike-Tcells 31 255
==> S79/nohup.out <==
aneuploid diploid
epi 46 53
ref-Bcells 313 80
ref-Tcells 28 449
spike-Bcells 183 57
spike-Tcells 31 258
==> S81/nohup.out <==
aneuploid diploid
epi 75 3
ref-Bcells 309 83
ref-Tcells 21 456
spike-Bcells 187 49
spike-Tcells 20 266
==> S82/nohup.out <==
aneuploid diploid
epi 70 15
ref-Bcells 83 308
ref-Tcells 455 22
spike-Bcells 49 186
spike-Tcells 259 27
This is very interesting. It can be seen that there are very strange discrimination of single cells in half of the patients, such as S82/nohup.out, in which a large number of ref and spike are judged as malignant cells. Let's look at its chart:
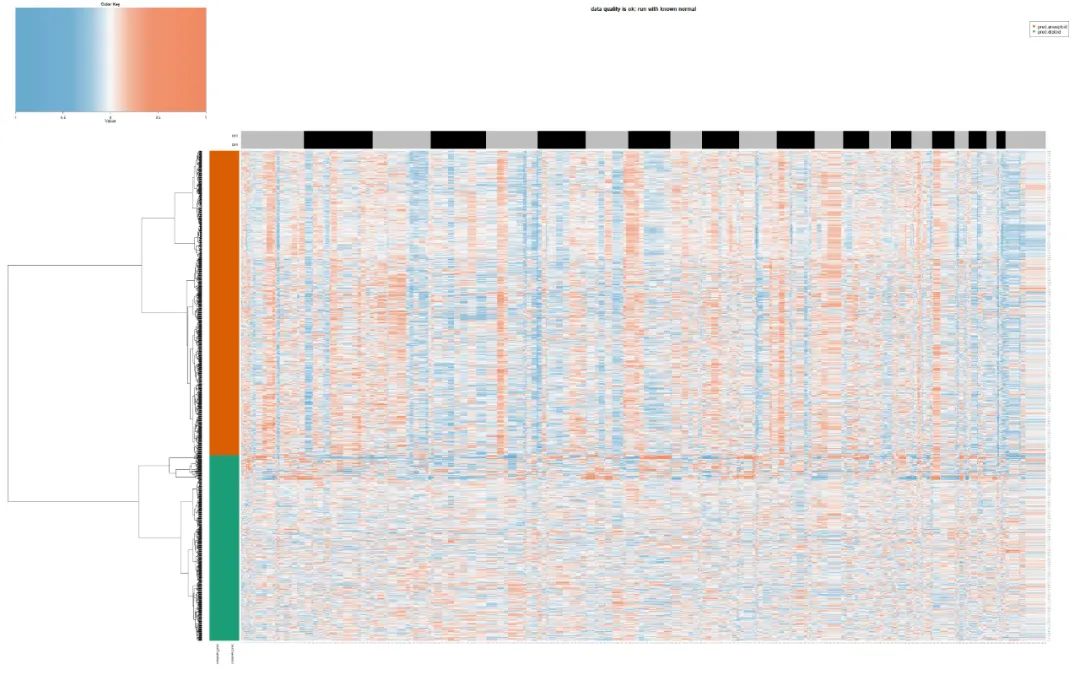
test_copykat_heatmap
It's really a little strange. At this time, the naked eye is actually the same as the software! And I went to see its inforcnv results, as shown below:
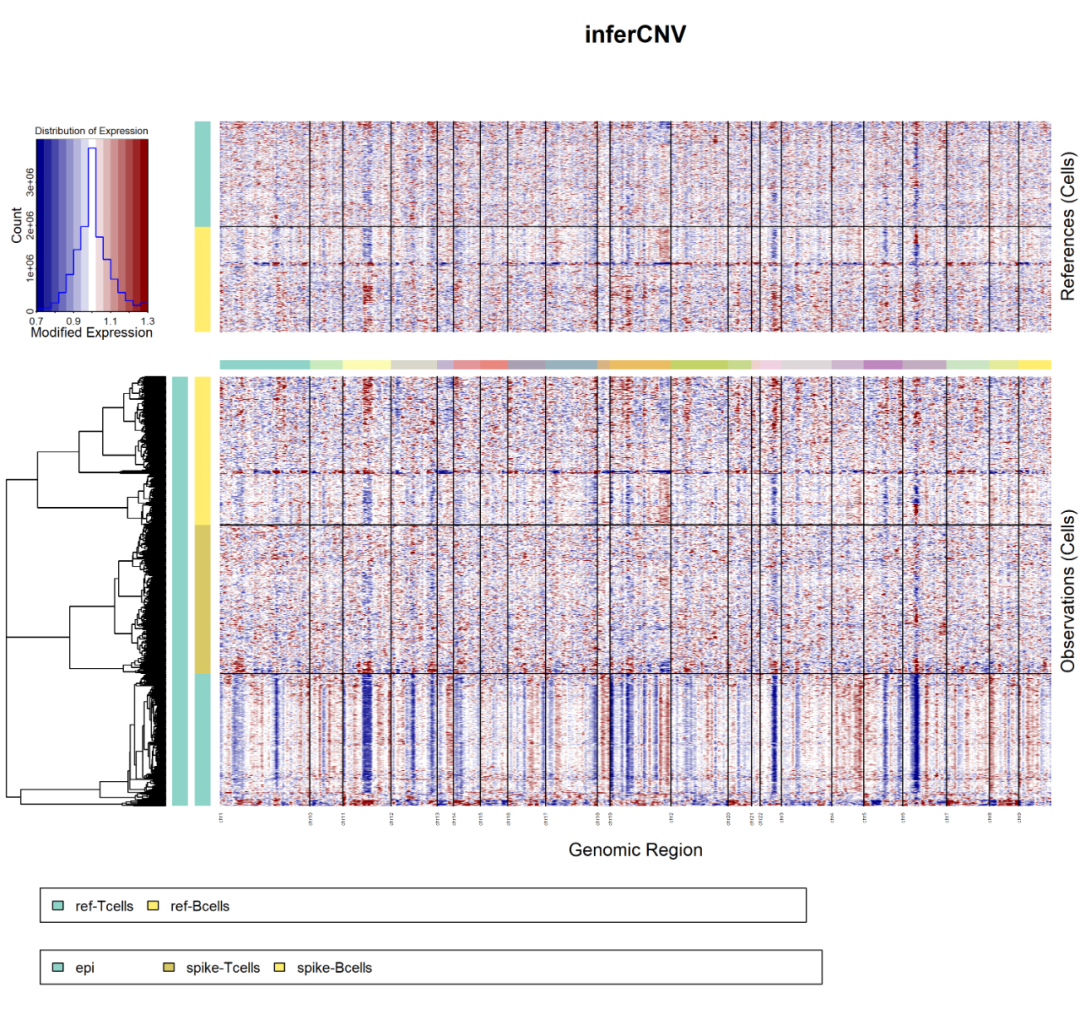
infercnv
It can be seen that copykat is just not intuitive, but in such patient data with a low proportion of malignant cells, there is no much difference in the effect, which is consistent with the conclusion of judging whether the cells are malignant or not by the naked eye! Although copykat mistakenly identified a large number of ref and spike as malignant cells, it is obvious that we will turn to the previous malignant epithelial cells as normal cells. At this time, there is some suspicion of idealism.
Although copykat is just not intuitive as inforcnv, copykat at least gives the judgment of aneuploid and diploid. The result file given by inforcnv still needs to be read by yourself, calculate the cnv score by yourself, and give a threshold to distinguish whether the cell is malignant or not. This step is also a test of your programming skills. You need to familiarize yourself with the following 5 tutorials:
- CNS chart reproduction 13 - use inforcnv to distinguish whether tumor cells are malignant or not
- CNS chart reproduction 14 - infocnv process for literature review
- CNS chart reproduction 15 - uncover the big difference of input data in infocnv process
- CNS chart reproduction 16 - Interpretation and utilization of infocnv results
- CNS chart reproduction 17 - advanced interpretation and utilization of infocnv results
If you also have your own copy number variation analysis needs of tumor related single-cell data, and you always report errors in many attempts, please leave a message and entrust us. The copy number variation single analysis of tumor related single-cell data is 2400. If the previous Dimension Reduction Clustering clustering is required, an additional 800 is required to provide an independent copykat and inforcnv process for distinguishing samples, as well as all the results and code folders of the combined copykat and inforcnv processes.
It should be noted that we will not keep the txt and other files in the middle, because it consumes too much disk space. For example, the project we demonstrated has less than 7000 epithelial cells from more than 20 patients. By following the copykat and inforcnv processes, we obtained nearly 100G files:
11G ./4-3-only-Epi 478M ./Rdata-celltype 81G ./epi-one-by-one