data sources
The transcriptome / genome of peripheral blood of Alpaca (seems to have been immunized) is amplified by multiple PCR to form a specific library, and these sequences are recombined into the expression vector and transferred into phage (phage display technology). After solid-phase / liquid-phase panning, a high affinity VHH sequence library is obtained. The sequence library is amplified again to form a high-throughput sequencing library, and PE300 sequencing strategy is adopted.
Experimental purpose
- paired reads assembled into productive conting
- Note contig obtains information such as FWR1/CDR1/FWR2/CDR2/FWR3/CDR3/FWR4
- Get clonotype
- Statistics of unique protein, unqiue clonotype, etc
MIXCR usage
Installing mixcr
wget https://github.com/milaboratory/mixcr/releases/download/v3.0.13/mixcr-3.0.13.zip #https://github.com/milaboratory/mixcr/releases unzip -d ~/software/mixcr mixcr-3.0.13.zip echo "export PATH=~/software/mixcr/bin/mixcr:$PATH" > ~/.bashrc source ~/.bashrc
overview
MIXCR of workflow It mainly includes three steps: 1,align: take reads And reference VDJC Isogenic comparison 2,assemble: utilize align Result assembly clonotype 3,export: take alignment perhaps clones Information export for 4,input: fasta/fastq/fastq.gz/paired-end fastq/paired_end fastq.gz 5,output:mixcr The result is a binary file that needs to be used exportAlignments and exportClones Export to readable format 6,Two packaged analysis modes: analyze amplicon for analysis of targeted TCR/IG library amplification (5'RACE, Amplicon, Multiplex, etc). analyze shotgun for analysis of random fragments (RNA-Seq, Exome-Seq, etc).
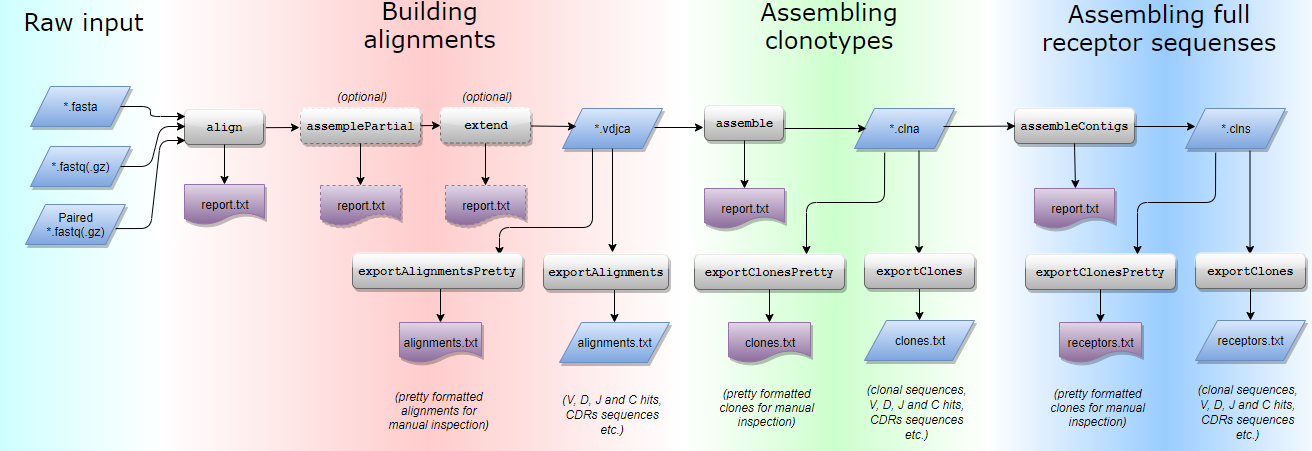
Example
Alpaca has no built-in reference sequence
MIXCR has built-in mouse and human reference
My data comes from alpaca, so I need to build the reference manually# https://mixcr.readthedocs.io/en/latest/importSegments.html
#Get josn file wget https://github.com/repseqio/library-imgt/releases/download/v6/imgt.201946-3.sv6.json.gz
Library search path: - built-in libraries - /home/username/. - /home/username/.mixcr/libraries - /software/mixcr/libraries
So I lost my josn file in / software/mixcr/libraries
#Specify reference library mixcr align --library imgt input_R1.fastq input_R2.fastq alignments.vdjca #Specifies the version of reference library mixcr align --library imgt.201631-4 input_R1.fastq input_R2.fastq alignments.vdjca
input
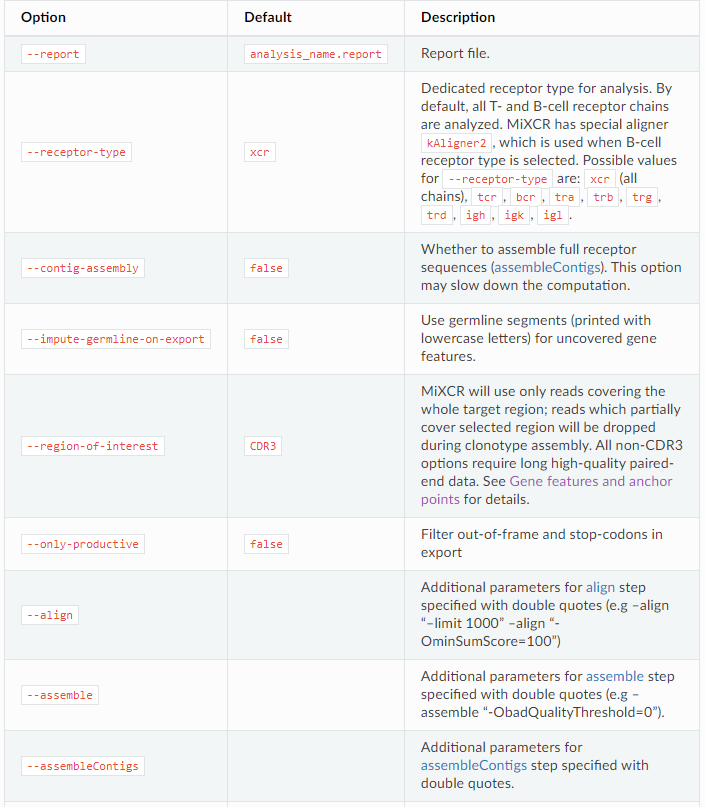
The software provides some parameters for controlling input
- – starting material: dna or rna for initial database building
- – 5-end: 5 'end primer no-v-primers or v-primers
- – 3-end: 3 'end primer j-primers or j-c-intron-primers or c-primers
- – adapters: is there a connector sequence? If so, it will help us do the trim action, adapters present or no adapters
output
. vdjca is a binary file generated by align
. clns is a binary generated by asemble
# export alignments from .vdjca file mixcr exportAlignments [options] alignments.vdjca alignments.txt # export alignments from .clna file mixcr exportAlignments [options] clonesAndAlignments.clna alignments.txt # export clones from .clns file mixcr exportClones [options] clones.clns clones.txt # export clones from .clna file mixcr exportClones [options] clonesAndAlignments.clna clones.txt #customize the list of fields that will be exported by passing parameters to export commands mixcr exportClones -count -vHit -jHit -vAlignment -jAlignment -aaFeature CDR3 clones.clns clones.txt
Analysis of targeted TCR/IG libraries
mixcr analyze amplicon -s alpaca \ #For the species of designated reference gene, BCR only uses human or mouse --starting-material rna \ #Amplification template used at the beginning of database construction --5-end v-primers --3-end j-primers \ #Amplification primers during library construction --adapters adapters-present \ #Are there any adapter s for sequencing or amplification primers for database construction --library imgt --receptor-type bcr \ #`tcr`, `bcr`, `tra`, `trb`, `trg`, `trd`, `igh`, `igk`, `igl` --contig-assembly \ #Do you want to assemble it contig #store initial reads in the resulting `.vdjca` file --only-productive ../NB244-R1-H_S1_L001_R1_001.fastq.gz ../NB244-R1-H_S1_L001_R2_001.fastq.gz \input analysis1 #prefix of output
–starting-material affects the choice of V gene region which will be used as target in align step (vParameters.geneFeatureToAlign, see align documentation): rna corresponds to the VTranscriptWithout5UTRWithP and dna to VGeneWithP (see Gene features and anchor points for details).
#In fact, vgenewithp = = {utr5begin: vent} + {vent: vent (- 20)}
VTranscriptWithout5UTRWithP == {L1Begin:L1End} + {L2Begin:VEnd} + {VEnd:VEnd(-20)}
#The generated files are as follows
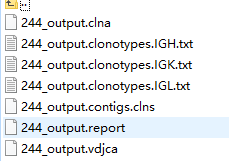

High quality full length IG repertoire analysis
mixcr analyze amplicon \
--species hs \
--starting-material rna \
--5-end v-primers \
--3-end j-primers \
--adapters adapters-present \
--receptor-type BCR \
--region-of-interest VDJRegion \
--only-productive \
--align "-OreadsLayout=Collinear" \
--assemble "-OseparateByC=true" \
--assemble "-OqualityAggregationType=Average" \
--assemble "-OclusteringFilter.specificMutationProbability=1E-5" \
--assemble "-OmaxBadPointsPercent=0" \
input_R1.fastq input_R2.fastq analysis2
##############################################################################################################################
#In the cluster step, we set searchdepth to 0. Are the VDJ sequences exactly the same
mixcr analyze amplicon \
--species hs \
--starting-material rna \
--5-end v-primers \
--3-end j-primers \
--adapters adapters-present \
--receptor-type BCR \
--region-of-interest VDJRegion \
--only-productive \
--align "-OreadsLayout=Collinear" \
--assemble "-OcloneClusteringParameters.searchDepth=0" \
--assemble "-OseparateByC=true" \
--assemble "-OqualityAggregationType=Average" \
--assemble "-OclusteringFilter.specificMutationProbability=1E-5" \
--assemble "-OmaxBadPointsPercent=0" \
input_R1.fastq input_R2.fastq analysis3
##############################################################################################################
problem
How is the clonotype of MIXCR defined?
After reading the instructions, I think the clonotype of MIXCR is defined as CDR3 NDA sequence, and those exactly the same are classified as a clonotype
The sequences after the cluster are not clonotype in our usual sense (same of V and j reference gene and similarity of cdr3_aa > = 80%)
Its cluster is also based on DNA sequence
mixcr assemble [options] alignments.vdjca output.clns #Building clonotype during assembly mixcr assemble [options] -a alignments.vdjca output.clna # the outputs result in a "clones & alignments" format, allowing subsequent contig assembly
The specific process is as follows:
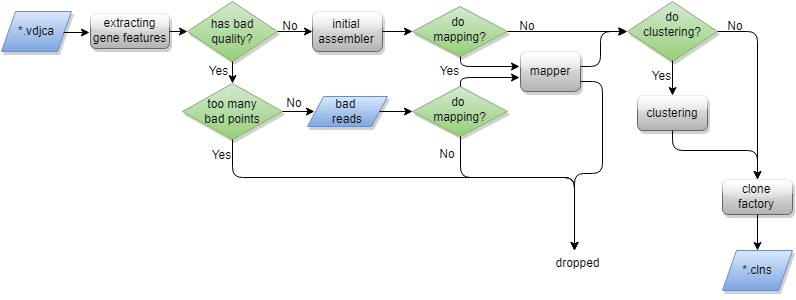
- Step 1: extract the clone sequence specified by assemblyfeatures parameter (CDR3 by default) from the alignment result file; If the aligned read does not contain a clone sequence, it will be discarded
- If the clonal sequence contains low-quality bases, it will be filtered according to badQualityThreshold and maxBadPointsPercent
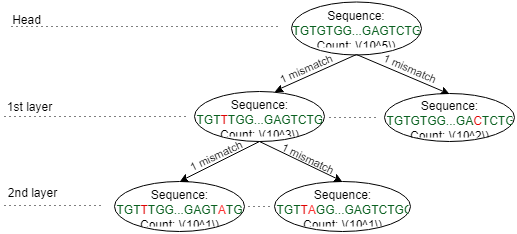
- After clonotypes are assembled by initial assembler and mapper, MiXCR proceeds to clustering. During clustering, clonotype with small counts will be attached to highly similar "parent" clonotypes with significantly greater count. After all clusters are built, only their heads are considered as final clones
Is alignment assembled first or compared first?
- Before PE-read alignment: overlap > 17bp,minimal identity, minimal fraction of matching nucleotides between sequences >=0.9
- After PE read alignment: when the merge threshold is not met, but the two reads are compared to the same V and J genes, start alignment aided overlaps to merge the reads
What if alignment encounters low-quality reads?
I didn't say it clearly or I didn't see it carefully, so it's best to do quality inspection and filtering when running mixcr
How to customize a clonotype?
One of the key MiXCR features is ability to assemble clonotypes by sequence of custom gene region (e.g. FR3+CDR3);
target clonal sequence can even be disjoint.
This region can be specified by assemblingFeatures parameter, as in the following example:
mixcr assemble -OassemblingFeatures="[V5UTR+L1+L2+FR1,FR3+CDR3]" alignments.vdjca output.clns
The control parameters of the assembly are as follows:

Separation of clones with same CDR3 (clonal sequence) but different V/J/C genes
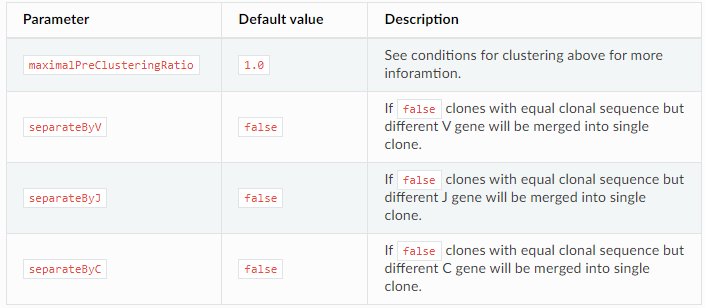
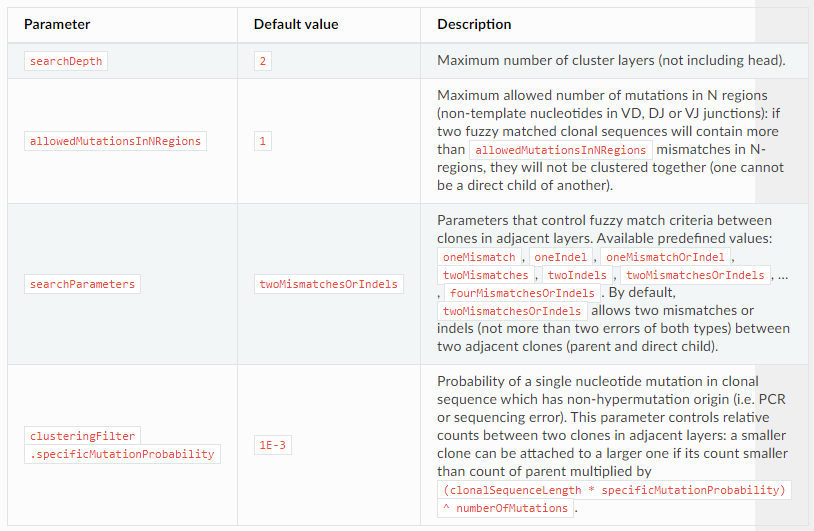
Clustering strategy: control clustering procedure are placed in cloneClusteringParameters parameters group which determines the rules for the frequency-based correction of PCR and sequencing errors:
How to understand assembly full TCR / Ig receiver sequences
Original text: mixcr allows to assemble full TCR / Ig receiver sequences (that is all available off-cdr3 regions) with the use of assemblyconstraints command. Full sequence assembly may be performed after building of initial alignments and assembly of ordinarycdr3 based clonotypes
Personal understanding: in MIXCR, assembly is assembly clones, which is the action of classifying the sequence of the same clonal sequence into a clonotype. Therefore, full receiver assembly should take the whole antibody sequence as a clonal sequence
https://mixcr.readthedocs.io/en/latest/assembleContigs.html
gene feature
The key feature of MiXCR is the possibility to specify:
- regions of reference V, D, J and C genes sequences that are used in alignment of raw reads
- regions of sequence to be exported by exportAlignments
- regions of sequence to use as clonal sequence in clone assembly
- regions of clonal sequences to be exported by exportClones
V Gene structure
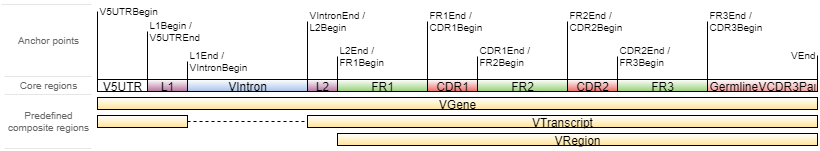
D Gene structure

J Gene structure

