Experiment 2: sequence assembly
1.art illumina simulated double terminal sequencing
Short and long insert libraries:
./art_illumina -ss HS25 -sam -i ./GCF_000146045.2_R64_genomic.fna -p -l 125 -f 10 -m 200 -s 10 -o ./Sc_paired ./art_illumina -ss HS25 -sam -i ./GCF_000146045.2_R64_genomic.fna -p -l 125 -f 10 -m 2500 -s 50 -o ./Sc_matepair
View results:
ll -o --block-size=M ./Sc_paired* ll -o --block-size=M ./Sc_matepair*
2. Create index file with bowtie2 build
bowtie2-build GCF_000146045.2_R64_genomic.fna Sc_index
Bowtie2 build changes the fasta file to the index database by default. $bowtie2 build < FASTA File > < prefix name of index file to survive >
3.fastqc quality control analysis
mkdir fastqc_out fastqc -o ./fastqc_out -f fastq -t 10 /usr/bin/art_bin_MountRainier/Sc_paired1.fq /usr/bin/art_bin_MountRainier/Sc_paired2.fq fastqc -o ./fastqc_out -f fastq -t 10 /usr/bin/art_bin_MountRainier/Sc_matepair2.fq /usr/bin/art_bin_MountRainier/Sc_matepair1.fq
4. Comparison between bowtie2 sequencing sequence and reference sequence
bowtie2 -x ./Sc_index -1 /usr/bin/art_bin_MountRainier/Sc_paired1.fq,/usr/bin/art_bin_MountRainier/Sc_matepair1.fq -2 /usr/bin/art_bin_MountRainier/Sc_paired2.fq,/usr/bin/art_bin_MountRainier/Sc_matepair2.fq -S ./Sc_2sets.sam -p 10
Generate. sam file

Double ended data comparison results:
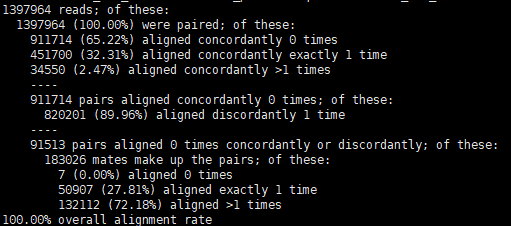
The first part describes the consistent alignment results under the pair end mode. aligned concordantly is that read1 and read2 are reasonably aligned to the genome / transcriptome at the same time.
The second part is the inconsistent comparison results in the pair end mode. concordantly 0 times is that read1 and read2 cannot be reasonably compared to the genome / transcriptome at the same time.
The third part is the comparison of the single ended modes of the remaining reads (neither concordantly nor discordantly 1 time).
5.samtools comparison results
It is a tool for processing alignment files in SAM/BAM (binary format of SAM, used to compress space) format. It can input and output files in SAM (sequence alignment/map) format, sort, merge and index them.
samtools view -b Sc_2sets.sam >Sc_2sets.bam #Format conversion Sam > BAM samtools sort Sc_2sets.bam -o Sc_2sets.sorted.bam #Sort by sequence name and output the results to Sc_2sets.sorted.bam samtools index Sc_2sets_sorted.bam #Indexing
6. Statistical analysis:
samtools stats ./Sc_2sets_sorted.bam > samtools.stat.stats.out samtools depth ./Sc_2sets_sorted.bam > samtools.stat.depth.out samtools flagstat ./Sc_2sets_sorted.bam > samtools.stat.flagstat.out samtools idxstats ./Sc_2sets_sorted.bam > samtools.stat.idxstats.out
7. Interpret the statistical results of samtools satas, and use plot bamstats to visualize the output results
plot-bamstats -p ./plot-bamstats_out/ ./samtools.stat.stats.out
Encountered error: missing: gunplot, download and install and rerun the above code
conda install -c bioconda gnuplot -y
8.SOAPdenovo-63mer sequence assembly and result analysis
nohup SOAPdenovo-63mer all -s lib.cfg -K 31 -o SOAPdenovo_out -p 10 & #Nohup & run in the background
An error is reported here because SOAPdenovo is not installed. Download it first
git clone https://github.com/aquaskyline/SOAPdenovo2.git cd SOAPdenovo2 make
An error occurred because Ubuntu 18.04.5 was used
Since 16.10, gcc has enabled the pie option by default. As a result, the mime of the compiled file is application/x-sharedlib. General file managers only recognize application/x-executable and do not treat it as an executable file
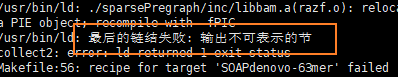
Modify gcc in Makefile
gcc -fno-pie -no-pie
Just make again
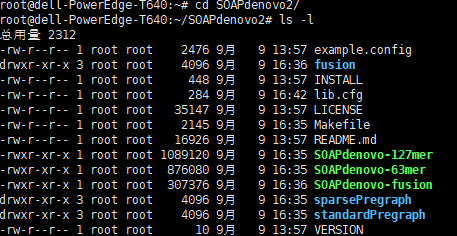
Due to the storage path of the following files, the configuration document needs to be modified
File path is
/usr/bin/art_bin_MountRainier
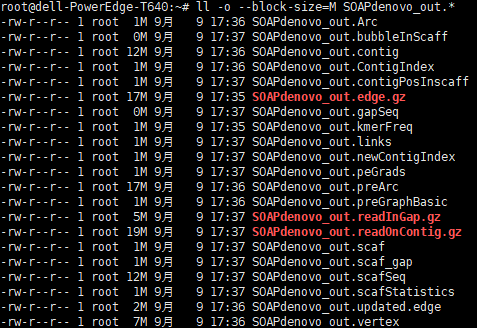
After changing a lot of problems, it must be possible to run. Try it, and it won't run in the background
SOAPdenovo2/SOAPdenovo-63mer all -s lib.cfg -K 31 -o SOAPdenovo_out -p 1
Yes, it's done. It's not easy for the spicy chicken
9.quast compares the documents of contings and scaffolds sequences in the assembly results with the reference genome respectively
quast-5.0.2/quast.py -o quast_out -r GCF_000146045.2_R64_genomic.fna -g GCF_000146045.2_R64_genomic.gff SOAPdenovo_out.contig #Contings assessment quast-5.0.2/quast.py -o quast_out -r GCF_000146045.2_R64_genomic.fna -g GCF_000146045.2_R64_genomic.gff SOAPdenovo_out.scafSeq #Scaffolding assessment
10. Let's save the results for free
Wuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwuwu